
Numer umowy: 2020/04/X/NZ9/00129
Przyznana kwota: 49 940 zł
Czas realizacji: 1.10.2020 – 25.08.2022
Cel projektu
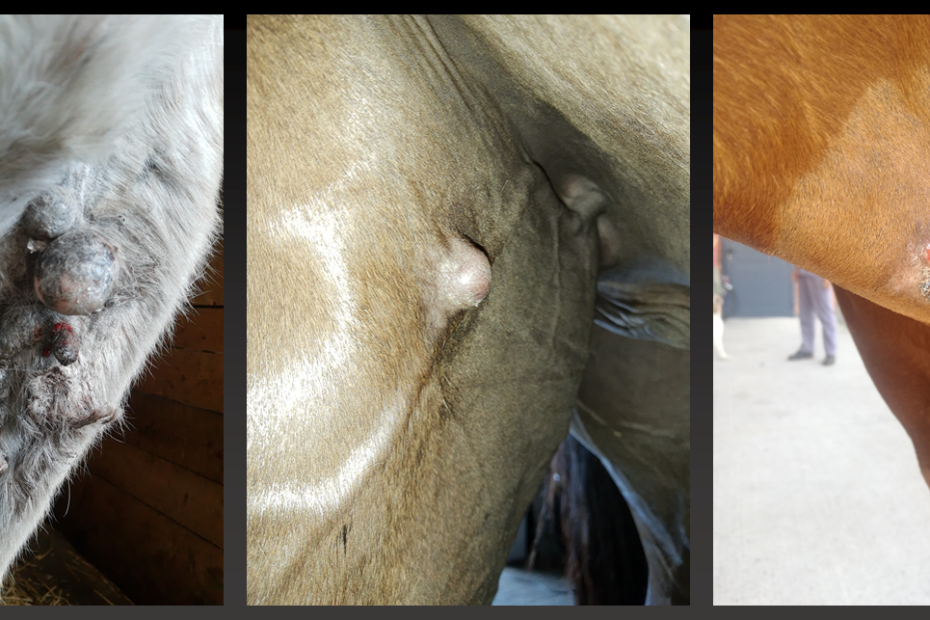
Zamierzeniem projektu jest porównawcza analiza metylacji genomu oraz identyfikacja genów kandydujących potencjalnie regulowanych przez aberrentną metylację DNA w trakcie patogenezy sarkoidu końskiego – najczęstszego typ nowotworu skóry u zwierząt koniowatych. Hipoteza badawcza zakłada, że nieprawidłowa ekspresja onkogenów i genów supresorowych w tym nowotworze może być wynikiem nieprawidłowej metylacji DNA, który może okazać się ważnym czynnikiem patogenezy sarkoidu.
Realizacja badań
- Do realizacji celu badań, wykorzystano technikę RRBS (ang. Reduced Representation Bisulfite Sequencing) i sekwencjonowanie następnej generacji NGS (ang. Next Generation Sequencing).
- Na podstawie analizy danych uzyskanych z NGS, stwierdzono wysoki współczynnik korelacji pomiędzy poziomem metylacji poszczególnych miejsc CpG w grupie tkanek nowotworowych i kontrolnych,, wskazujący na duże podobieństwo globalnego wzorca metylacji DNA pomiędzy badanymi próbami.
- Łącznie zidentyfikowano 29 311 różnicowo metylowanych miejsc (DMS) i 11 696 regionów (DMR) genomu; największą liczbę DMS i DMR zaobserwowano w intronach genów, a następnie w regionach międzygenowych i sekwencjach kodujących genu.
- Analiza przeprowadzana dla wszystkich genów w obrębie których zidentyfikowano DMS, niezależnie od poziomu ekspresji, wskazała na istnienie wysoce istotnej, negatywnej korelacji (rho=-0,328; p=4,132e -7) pomiędzy zmianami w metylacji promotorów genów a zmianami w ich ekspresji w tkance sarkoidu. Podobną zależność, lecz o nieco mniejszym nasileniu (rho=-0,126; p=5,280e -21), zaobserwowano dla zmian w metylacji ciał analizowanych genów,
- W badaniach zidentyfikowano także geny potencjalnie deregulowane przez aberrentną metylację w sarkoidzie; dla DMS zlokalizowanych w promotorach zidentyfikowano 12 genów wykazujących prawidłową, negatywną zależność metylacji i ekspresji (w tym POPDC2, SFN, PRNP, LPAR1, CALB2, MPZL3 i TRIM29). Większość z tych genów (n=8), obejmowała DMS charakteryzujące podwyższonym poziomem metylacji i obniżoną ekspresją (adjp<0.05). W przypadku DMS zlokalizowanych w ciałach genów, 111 genów (powiązanych z 268 DMS) wykazało istotne zmiany w poziomie ekspresji. Niemniej jednak, zależność pomiędzy metylacją i ekspresją miała niejednorodny charakter.
- Analiza umożliwiła wytypowanie 12 białek, których wiązanie do nici DNA może potencjalnie ulegać zaburzeniom na skutek metylacji analizowanych miejsc CpG (w tym R2, TGA1a, HES-1, Bcd, HSF1 i E74A).
Przeprowadzone analizy umożliwiły scharakteryzowanie profilu metylacji DNA oraz identyfikację genów kandydujących potencjalnie regulowanych przez metylację DNA w patogenezie sarkoidu. Uzyskane wyniki stanowią bazę do dalszych badań zmierzających do identyfikacji potencjalnych markerów molekularnych, umożliwiających diagnostykę tego schorzenia u koni.
Publikacje
- Semik-Gurgul E., Szmatoła T., Gurgul A., Pawlina-Tyszko K., Gałuszka A., Pędziwiatr R., Witkowski M., Ząbek T. (2023). Methylome and transcriptome data integration reveals aberrantly regulated genes in equine sarcoids. Biochimie, 13:100-113. doi: 10.1016/j.biochi.2023.05.008.
- Semik-Gurgul E., Gurgul A., Pawlina-Tyszko K., Gałuszka A., Pędziwiatr R., Witkowski M., Ząbek T. (2023). Involvement of aberrant DNA methylation in the deregulated expression of EHF, LPAR1, MPZL3, and POPDC2 genes in equine sarcoids. Annals of Animal Science. 24, 99–107. DOI: 10.2478/aoas-2023-0078
- Semik-Gurgul E., Gurgul A., Szmatoła T. (2023). Transcriptome and methylome sequencing reveals altered long non-coding RNA genes expression and their aberrant DNA methylation in equine sarcoids. Functional & Integrative Genomics, 8;23(3):268. doi: 10.1007/s10142-023-01200-2.